
Newly-Detected Solitary Bony Lytic/Sclerotic Lesion with Soft Tissue Mass in a Previously Treated Case of High-Risk Medulloblastoma: Importance of Contemporary Pathology Techniques to Differentiate Second Malignant Neoplasm from Extra-Neuraxial Metastasis
Abstract
Multi-modality therapy has led to significant improvement in outcomes for childhood medulloblastoma; however, long-term survivors have become more susceptible to late effects of therapy including induction of second malignant neoplasms and even remain at an increased risk of late relapses including extra-neuraxial metastases. A newly detected solitary lytic/sclerotic osseous lesion in a medulloblastoma survivor away from the radiation field poses considerable diagnostic challenge as it could represent either a second malignant neoplasm or extra-neuraxial metastasis. We report one such case highlighting the importance of contemporary pathology techniques as useful adjuncts to differentiate a second primary osseous Ewing’s sarcoma (ES)/primitive neuro-ectodermal tumor (PNET) from bony metastasis and review the pertinent literature on second malignant neoplasms and extra-neuraxial metastases in medulloblastoma. To the best of our knowledge, this is the first report of a molecularly confirmed second primary osseous ES/PNET in a survivor of childhood medulloblastoma.
Author Contributions
Academic Editor: Pier Paolo Panciani, Department of Neuroscience, Division of Neurosurgery, University of Torino and Brescia (Italy)
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2015 Tejpal Gupta, et al
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interests:
The authors have declared that no competing interests exist.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interests:
The authors have declared that no competing interests exist.
Citation:
Introduction:
Medulloblastoma, the most common pediatric malignant brain tumor is a molecularly heterogeneous disease with different developmental origins, distinct phenotypes, diverse clinical behavior and contrasting clinical outcomes.1 Current multi-disciplinary therapy has led to significant improvement in survival for childhood medulloblastoma. With progressive improvements in survival, second malignant neoplasms (SMN) although rare, are now being increasingly reported in patients with medulloblastoma.2 At the same time, extra-neuraxial metastases (ENM), though uncommon, are also being detected increasingly3 particularly in patients with high-risk disease. The diagnosis is relatively straightforward in the presence of multiple skeletal lesions or marrow involvement. However, a newly-detected solitary lytic/sclerotic osseous lesion in a medulloblastoma survivor poses considerable diagnostic challenge. The differential diagnoses in such an instance is between a second primary osseous Ewing’s family tumor such as Ewing’s sarcoma (ES)/primitive neuro-ectodermal tumor (PNET) and bony metastasis in a known case of medulloblastoma. Both entities look rather similar (small blue round cell tumor) on light microscopy mandating appropriate contemporary pathology techniques such as immunohistochemistry (IHC) and molecular biology tests for confirmation. We report one such case and review the pertinent literature on SNM and ENM in medulloblastoma.
Clinical Summary:
A 6-year old boy initially presented to us with headache, vomiting, and ataxia of 6-months duration. Cranial magnetic resonance imaging (MRI) showed a large lobulated, midline vermian space-occupying lesion, hypo-to iso-intense on T1-weighted images, hyperintense on T2-weighted and FLAIR images with prominent and homogenous enhancement post-contrast (Figure 1A-C) with aqueductal compression causing supratentorial hydrocephalus suggestive of medulloblastoma. He underwent near-total excision of the vermian space occupying lesion at an outside hospital that was reported as classical medulloblastoma on conventional light microscopy (Figure 1D). The non-availability of formalin-fixed paraffin embedded tumor tissue blocks precluded further molecular subgrouping of medulloblastoma. Post-operative MRI revealed the presence of two small residual enhancing nodules at the edge of the resection cavity. Neuraxial staging using MRI of the spine with gadolinium and cerebrospinal fluid (CSF) malignant cell cytology via a lumbar puncture did not show any evidence of leptomeningeal dissemination. Although there was no evidence of leptomeningeal metastases (M0 status), he was categorized as high-risk disease by virtue of residual tumor volume >1.5 X 1.5cm2 as per the prevalent risk-stratification system, and treated on an ongoing institutional phase II study of concurrent carboplatin with standard-dose craniospinal irradiation with posterior fossa boost followed by six cycle of multi-agent adjuvant systemic chemotherapy. Neuraxial imaging after completion of adjuvant therapy showed complete resolution of residual enhancing nodules with reactive gliosis.
Figure 1.Pre-operative MRI of the brain showing a brilliantly enhancing midline vermian lesion in axial (A) and sagittal (B) T1-weighted post-contrast images with variable intensity on T2-weighted images (C). Photomicrograph of the tumor (D) showing it highly cellular tumor composed of small blue round cells consistent with classic medulloblastoma (X 400, hematoxylin & eosin)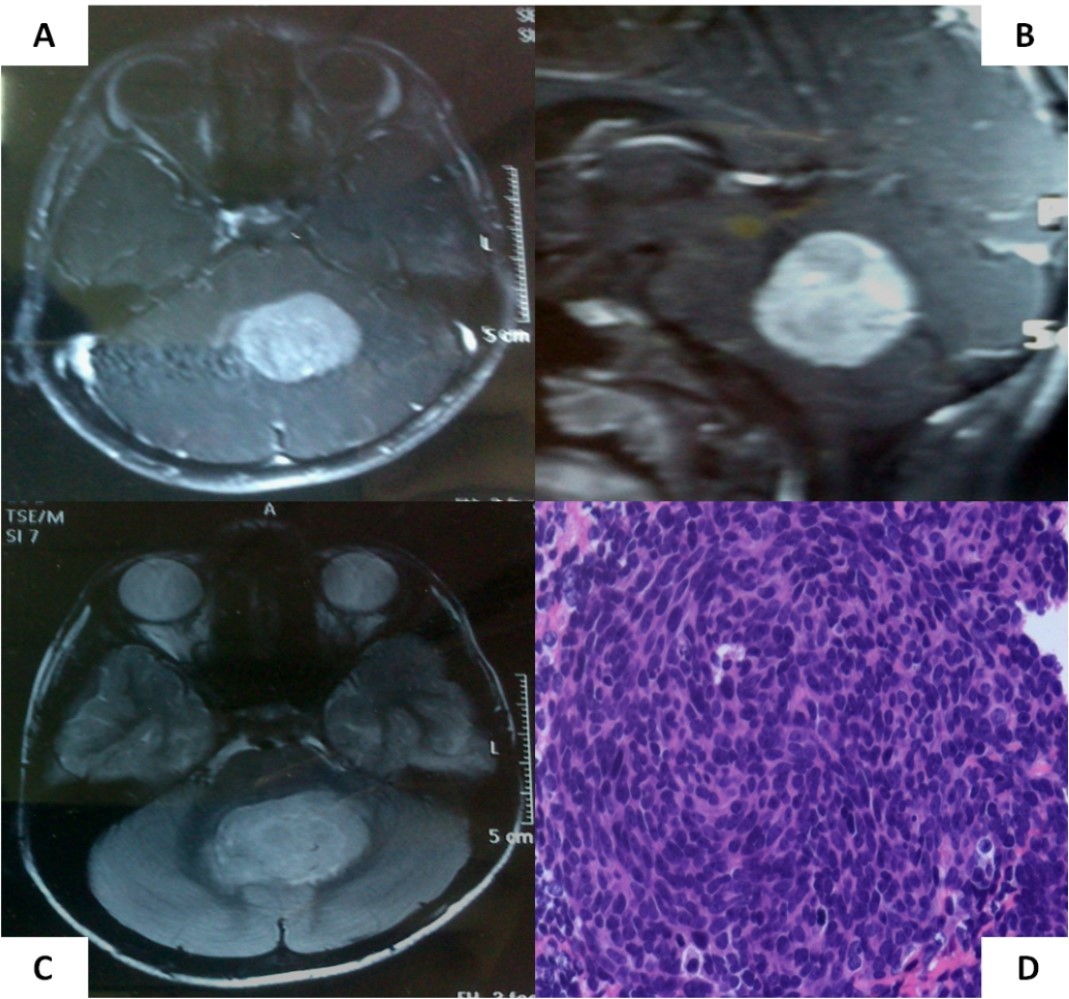
Two and half years after completion of therapy (just over 3 years from initial diagnosis), the child presented with pain in the left lower limb following a trivial injury. Local radiograph showed a large lytic/sclerotic lesion in the proximal left tibia. MRI of the left lower extremity (Figure 2A-B) confirmed the presence of this lytic/sclerotic lesion arising from the proximal tibial metaphysis with significant invasion of the adjacent soft tissues. The lesion was isointense on T1-weighted images, heterogeneously hypointense on T2-weighted images, hyperintense on STIR images with moderate post-contrast enhancement. Whole-body 18-F-fluoro-deoxy-glucose positron emission tomography/computed tomography (FDG-PET/CT) showed increased radiotracer uptake limited to the mass lesion in the proximal left tibia with a maximum standardized uptake value (SUVmax) of 6.28 suggestive of localized metabolically active tumor. There were no other areas of abnormal tracer uptake anywhere in the body ruling out disseminated disease (Figure 2C). Neuraxial imaging at this time did not reveal any local recurrence in the posterior fossa or leptomeninges confirming that the index cancer was controlled. The differential diagnosis was between second primary bone neoplasm with extensive soft tissue component versus solitary metastasis from medulloblastoma.
Figure 2.Axial (A) and coronal (B) MRI sections of left proximal tibia showing a large soft tissue component in addition to the lytic/sclerotic bony lesion. Whole body FDG-PET/CT showing increased tracer uptake localized to this lesion (C) with no significant abnormal uptake elsewhere in the body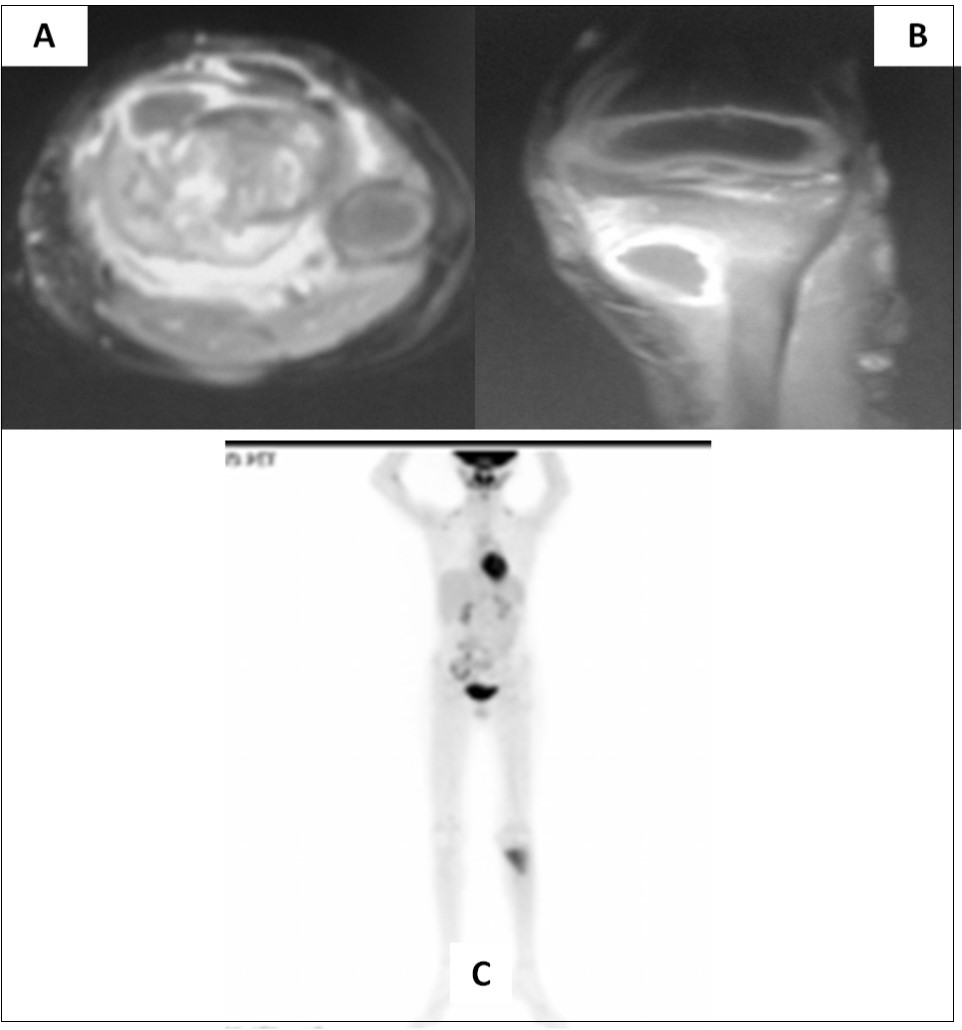
Pathological findings:
A J-needle biopsy from the extremity tumor showed the tumor to be composed of ‘undifferentiated small blue round cells’ (Figure 3A) with variable cytoplasmic vacuolation. To characterize the lesion further, appropriate special stains and IHC was carried out on the biopsy specimen. The vacuolated cells stained positive for Periodic Acid Schiff (Figure 3B) while the small blue round cells were diffusely membrane positive for CD99 or Mic2 (Figure 3C), focally weak positive for FLI-1, and negative for LCA and desmin suggesting a primary ES/PNET rather than metastases from medulloblastoma. To aid in definitive diagnosis, molecular testing was done showing Ewing’s sarcoma breakpoint region 1 (EWSR1) gene re-arrangement (Figure 4) on fluorescence in-situ hybridization (FISH). Ewing’s sarcoma is strongly associated with translocation t(11;22) in most cases4 and t(7;22) or t(21;22) in some cases, all involving the EWSR1 gene on chromosome 22.
Figure 3.Biopsy from the tibial lesion (A) showed ‘undifferentiated small blue round cells’ on light microscopy (X400, hematoxylin & eosin). Vacuolated cells were positive for Periodic Acid Schiff stain (B), while small blue round cells showed diffuse membrane positivity for Mic-2 (C)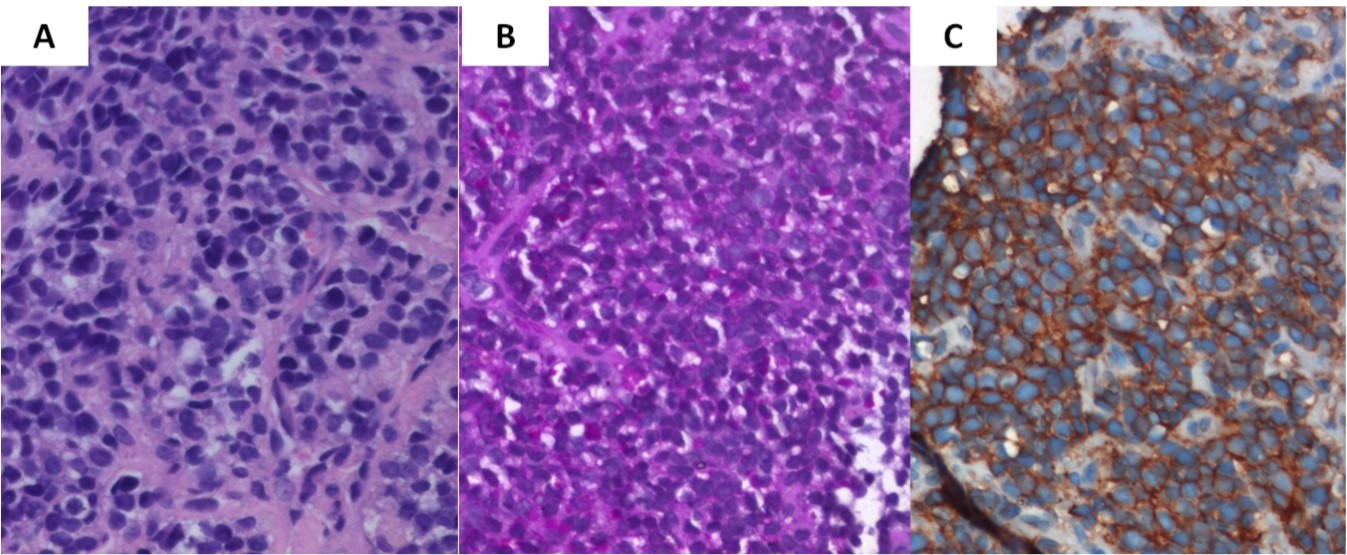
Figure 4.Representative photomicrograph of interphase fluorescence in-situ hybridisation (FISH) for Ewing’s sarcoma breakpoint region 1 (Vysis EWSR1 dual colour break apart kit; Abbott Molecular Inc.) showing spilt red and green signals (as shown by the encircled circles) indicating EWSR1 gene re-arrangement strongly supportive of a diagnosis of Ewing’s sarcoma
Therapy and Follow-Up:
Based on the diagnosis of localized ES/PNET of the lower extremity, the child was treated with a short intensive induction chemotherapy regimen (8-weeks) comprising 2 cycles each of vincristine, ifosfamide, etoposide (VIE) and vincristine adriamycin, cyclophosphamide (VAC) given sequentially followed by definitive local radiotherapy to the left tibia resulting in significant resolution of the soft tissue mass and stable bony changes in the proximal tibia. Maintenance chemotherapy following local radiotherapy consisted of 2 cycles of VIE chemotherapy followed 6 cycles of alternating vincristine, cyclophosphamide, actinomycin-D (VCD) and VAC chemotherapy lasting for 6 months. Nearly one year after completion of chemo-radiotherapy for the second primary ES/PNET, the child was detected with widespread metastases (lungs, bones, bone marrow) during clinico-radiological surveillance on follow-up, to which he succumbed shortly thereafter.
Discussion:
Incremental advances in neuro-surgical techniques, improved radiotherapy planning and delivery and newer chemotherapeutic agents have consistently and progressively improved the prognosis for medulloblastoma, a highly cellular, embryonal tumor of the central nervous system (CNS) arising in the cerebellum with inherent propensity of CSF dissemination. The long-term survival in average-risk medulloblastoma exceeds 85%5 and reaches 60-70% even for patients with high-risk disease,6 making it a success story of contemporary pediatric neuro-oncology. With such improvements in survival, children with medulloblastoma have becoming more susceptible to late effects of therapy including induction of SMN as well as remain at an increased risk of late relapses including ENM. In long-term survivors of childhood medulloblastoma, there is nearly four-fold excess risk of developing a SMN compared to the general population. (Table 1)2, 5, 7, 8. These occur typically several years after the diagnosis and treatment of the index cancer and include brain tumors (gliomas, meningiomas), thyroid cancers, sarcomas, and leukemias. Majority of solid second cancers occur originate either within or at the periphery of radiotherapy portals suggesting a possible causal association with radiation.
ENM in medulloblastoma (Table 1)3, 9, 10, 11, though uncommon, is reported in <1% patients at initial diagnosis and about 5-10% of patients during the course of their disease. Bone is the most common site of systemic involvement followed by bone marrow, lymph nodes, lung, and liver. Ventriculo-peritoneal shunts increase the risk of extraneural spread, particularly to the abdomen and lymph nodes. ENM generally occurs few months to years after initial diagnosis and is nearly always fatal despite aggressive treatment. Concurrent CNS involvement, lung or liver involvement, age <16 years, and time interval <18 months from initial diagnosis to ENM are negative prognostic factors for survival.10 A recent report11 indicates that desmoplastic variants may be more prone to develop systemic metastases which may be compatible with longer survival suggesting that pathologic subtype and molecular subgroup may play a role in determining outcome.
Table 1. Selected studies reporting second malignant neoplasms (SMN) and extraneural metastases (ENM) in medulloblastoma| Author (ref) | Number of patients | Patients with event | Risk estimates/Proportion 95% confidence interval (CI) | Median time (range) to onset of event of interest | Common type/site of event | |
| Studies reporting second malignant neoplasms | ||||||
| Goldstein (2) | 1262 | 20 | 5.4 fold (95%CI=3.3-8.4) excess risk of SMN | 73 months (8-432 months) | Cancers of the salivary gland, cervix, thyroid, glioma and acute lymphoblastic leukemia | |
| Packer (5) | 379 | 15 | Four-fold excess risk of SMN. Cumulative 10-year incidence: 4.2% (95%CI=1.9-6.5%) | 67 months (37-201 months) | High-grade glioma, thyroid cancer, myelodysplasia, acute leukemia, and osteosarcoma | |
| #Armstrong (7) | 1877 | 76 | Four-fold excess risk of SMN. Standardized Incidence Ratio: 4.1 (95%CI=3.2-4.2) | 192 months (61-319 months) | High-grade glioma, thyroid cancer, meningioma and soft tissue sarcoma | |
| Strodtbeck (8) | 1338 | 65 | Relative Risk: 4.31 (95%CI=3.33-5.49) | Risk increased in all latency periods | Cancer of digestive system, urinary system, brain, bone/joint, thyroid and leukemia | |
| Studies reporting extraneural metastases | ||||||
| Rochkind (3) | Not known | 89 children30 adults | Overall mean rate of 7.1%(range 1-20%) | Children: 20 months (1-76months)Adults: 36 months (2-120months) | Bones, lymph nodes, lungs, liver and soft tissues | |
| Kochbati (9) | 103 | 8 | 7.7% rate of ENM | 23 months (8-53 months) | Bones and lymph nodes | |
| Mazloom (10) | Not known | 119 | Not reported | 16 months (0-164 months) | Bones, bone marrow, lymph nodes, lungs and liver | |
| Young (11) | 292 | 14 | 4.8% rate of ENM | 18 months (2-208 months) | Bones and bone marrow | |
The time to development of SMN from index cancer diagnosis in our case was earlier than expected (36 months only). The possible cause of ES/PNET in our patient remains speculative. There was neither a significant family history of cancer nor did he have any known genetic predisposition syndrome to cancer. The site of second primary (proximal tibia) in our patient was considerably away from the caudal edge of the spinal radiotherapy field thereby ruling out the chance of it being a radiation-induced sarcoma. Secondary cancers following chemotherapy12 mostly involve the hematopoetic system (leukemia and myelodysplasia), although some alkylating agents such as cyclophosphamide have also been associated with development of solid tumors as well. The use of cyclophosphamide for the treatment of medulloblastoma could be one such potential association with metachronous ES/PNET in our patient.
The implications of potential misdiagnosis of ENM vis-à-vis SMN cannot be overemphasized. Systemic metastases from medulloblastoma is generally associated with an extremely poor prognosis with median survival rarely exceeding 6 months despite aggressive treatment in contrast to metachronous second primary tumor that may be amenable to long-term survival depending upon the histologic type and grade of the SMN as well as therapeutic management. Therefore, it becomes imperative that appropriate histopathological and molecular biology tests be performed to provide a definitive diagnosis.
To the best of our knowledge, this is the first report of a molecularly confirmed second primary osseous ES/PNET in a survivor of childhood medulloblastoma. An extensive and systematic search of the indexed medical literature failed to identify any previous such report. We did identify one recent report13 of recurrent extra-neural metastatic medulloblastoma in an adult who presented with progressive bilateral cervical lymphadenopathy and a large soft tissue mass associated with the right suboccipital craniotomy scar, 3-years after prior therapy for lateralized cerebellar medulloblastoma. He achieved near complete response to 4 cycles of ES/PNET type chemotherapy (vincristine, adriamycin, cyclophosphamide alternating with ifosfamide and etoposide). Subsequently he underwent high-dose chemotherapy (conditioning regimen of thiotepa, etoposide and carboplatin) with autologous hematopoetic stem-cell transplantation and local radiotherapy to the occipito-cervical region as consolidation and remained in durable remission for over 30 months after salvage therapy.
Conclusion:
A newly-detected solitary lytic/sclerotic osseous lesion in a medulloblastoma survivor poses considerable diagnostic challenge as it could represent either a SMN or ENM. Time course from index cancer diagnosis and presence of other sites of disease may help resolve this dilemma to a large extent. Appropriate histopathological, immunohistochemical, and molecular biology tests are useful adjuncts that aid in definitive diagnosis.
Funding:
No source of funding was involved in preparation of the manuscript
Acknowledgements
None
References
- 1.Taylor M D, Northcott P A, Korshunov A, Remke M, Cho Y et al. (2012) Molecular subgroups of medulloblastoma: the current consensus. , Acta Neuropathol 123, 465-472.
- 2.Goldstein A M, Yuen J, Tucker M A. (1997) Second cancers after medulloblastoma: population-based results from the United States and Sweden. , Cancer Causes Control 8(6), 865-871.
- 3.Rochkind S, Blatt I, Sadeh M, Goldhammer Y. (1991) Extracranial metastases of medulloblastoma in adults: literature review. , J Neurol Neurosurg Psychiatry 54(1), 80-86.
- 4.Turc-Carel C, Aurias A, Mugneret F, Lizard S, Sidaner I et al. (1988) Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of,t(11;22)(q24;q12).Genet Cytogenet. 32(2), 229-238.
- 5.Packer R J, Zhou T, Holmes E, Vezina G, Gajjar A. (2013) Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children’s Oncology Group trial A9961. , Neuro-Oncol 15(1), 97-103.
- 6.Jakacki R I, Burger P C, Zhou T, Holmes E J, Kocak M et al. (2012) Outcome of children with metastatic medulloblastoma treated with carboplatin during craniospinal radiotherapy: a Children’s Oncology Group Phase I/II study. , J Clin Oncol 30(21), 2648-653.
- 7.Armstrong G T, Liu Q, Yasui Y, Huang S, Ness K K et al. (2009) Long-term outcomes among adult survivors of childhood central nervous system malignancies in the Childhood Cancer Survivor Study. , J Natl Cancer Inst 101(13), 946-958.
- 8.Strodtbeck K, Sloan A, Rogers L, Fisher P G, Stearns D et al. (2013) Risk of subsequent cancer following a primary CNS tumor. , J Neurooncol 112(2), 285-295.
- 9.Kochbati L, Bouaouina N, Hentati D, Nasr C, Besbes M et al. (2006) Medulloblastoma with extracentral nervous system metastases: clinical presentation and risk factors. , Cancer Radiother 10(3), 107-111.
- 10.Mazloom A, Zangeneh A H, Paulino A C. (2010) Prognostic factors after extraneural metastasis of medulloblastoma. , Int J Radiat Oncol Biol Phys 78(1), 72-78.
- 11.Young R J, Khakoo Y, Yhu S, Wolden S, De Braganca KC et al. (2015) Extraneural metastases of medulloblastoma: Desmoplastic variants may have prolonged survival. , Pediatr Blood Cancer 64(4), 611-615.
- 12.Boffetta P, Kaldor J M. (1994) Secondaries malignancies following cancer chemotherapy. , Acta Oncol 33(6), 591-598.
- 13.Clement J, Varlotto J, Rybka W, Frauenhoffer E, Drabick J J. (2013) Unusual case of recurrent extraneural metastatic medulloblastoma in a young adult: durable complete remission with Ewing sarcomachemotherapy regimen and consolidation with autologous bone marrow transplantation and local radiation. , J Clin Oncol 31(19), 316-319.